

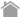
新潟大学 小松 雅明 先生・一村 義信 先生より
『p62/Sqstm1:オートファジーとKeap1-Nrf2システム
を繋ぐ分子』

|
 |
|||||||
| 小松 雅明 先生 新潟大学大学院医歯学総合研究科 分子細胞生物学 |
一村 義信 先生 新潟大学大学院医歯学総合研究科 分子細胞生物学 |
p62/Sqstm1:オートファジーとKeap1-Nrf2システムを繋ぐ分子
オートファジーとKeap1-Nrf2システムは、環境ストレスに応じて発動する細胞防御機構である。前者は細胞毒性を持つ細胞内成分をリソソームで分解することで、後者は抗酸化タンパク質の遺伝子発現を誘導することで細胞を保護する。二つの経路は共通のストレスにより誘導されるが、その相互関連は不明であった。最近、Nrf2の標的遺伝子産物でありオートファジーによって選択的に分解されるp62/Sqstm1が、両経路を連携させるキー分子であることが明らかになった。
はじめに
p62/Sqstm1(以降はp62と省略)はPhox and Bem1p(PB1)ドメイン、ジンクフィンガー、2つの核内移行シグナル、TRAF6結合ドメイン、核外移行シグナル、LC3相互作用領域(LIR)、Keap1相互作用領域(KIR)そしてユビキチン会合(UBA)ドメインを有するストレス誘導性の多機能タンパク質である[1, 2](図1)。この後生動物に保存されたタンパク質はその発見以来atypical PKC、ERK1、NF- κB、 caspase-8およびmTORC1のシグナリングアダプターとして解析されてきた。p62が広く注目されるようになったのは、2005年にJohansenらによりp62がユビキチン化凝集体をオートファゴソームへと導くレセプターとして機能することを提唱してからである[3]。2007年に著者らはp62が直接オートファゴソーム局在タンパク質LC3と相互作用すること、そしてオートファジー欠損マウス組織においてp62は顕著に蓄積しユビキチン化タンパク質とともに凝集化することを報告した[4]。さらに、2007年にJohansenらの生化学的解析から、2008年には著者らの構造解析からp62はDDDWTHL配列(LIR)を介してLC3と結合することが判明した[5, 6]。 現在、多くのオートファジーレセプターやLC3/Atg8結合タンパク質が持つLIRの先駆的研究としてp62のドメインおよび構造解析が貢献した。さらに、その後p62は脱分極したミトコンドリアや細胞内侵入細菌などをユビキチン鎖により認識し、それら積み荷(カーゴ)をオートファゴソームに運ぶレセプターとしての機能も続々と報告された[7]。2010年には、著者、Zhangらのグループが独立に、p62がCullin3型ユビキチンリガーゼのアダプタータンパク質Keap1と直接に相互作用し、酸化ストレス応答のマスター転写因子であるNrf2を活性化する仕組みが明らかになった[8, 9]。さらに、Johansenらはp62がNrf2の標的遺伝子の一つであることを示し、p62-Keap-Nrf2にポジティブフィードバック機構があることを示した[10]。2005年以降、p62はオートファジーそして酸化ストレス応答の表舞台に立ちつづけている。ここでは、現時点におけるp62のオートファジーレセプター機能と連結したNrf2活性化機構を述べる。p62のシグナリングハブとしての機能等は他の総説を参照いただきたい[11, 12]。
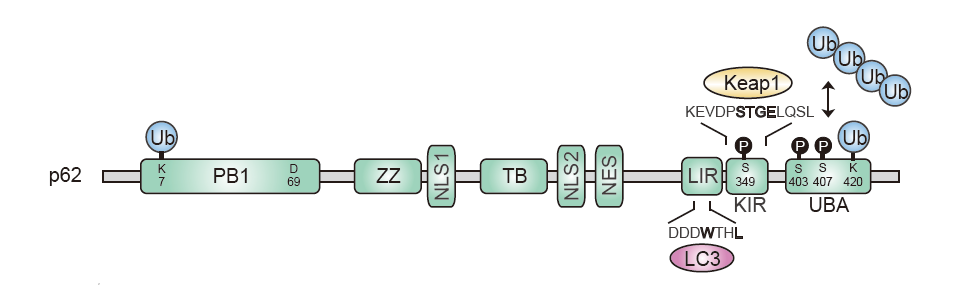
図1. p62ドメイン構造
p62はPhox and Bem1p(PB1)ドメイン内の7番目のリシン残基、69番目のアスパラギン酸残基を介して多量体を形成し、弾力性のあるらせん状フィラメントを形成する。ユビキチンリガーゼTRIM21による7番目のリシン残基のユビキチン化は多量体形成を阻害する。LC3-interacting region(LIR)の340番目のトリプトファンと343番目のロイシンがLC3やGABARAPの2つの疎水性ポケットと疎水性相互作用を形成する。C末端のUbiquitin-associated(UBA)ドメインに存在する407番目のセリン残基および403番目のセリン残基のリン酸化が、それぞれUBAドメインの構造変換およびユビキチン鎖との親和性を高める。420番目のリシン残基のユビキチン化は二量体形成を妨げる。Keap1-interacting region(KIR)はKeap1と結合する。349番から352番目のSTGE配列を介してKeap1のβ-プロペラ構造の底面に結合する。349番目のセリン残基がリン酸化されることによりKIRのKeap1への親和性は30倍以上高まる。
ZZ:zinc finger、TB:TRAF6-binding domain、NLS:核移行シグナル、NES:核外移行シグナル。
1.選択的オートファジーレセプターとしてのp62
p62のN末端に存在するPB1ドメインは塩基性アミノ酸クラスターと酸性アミノ酸からなるOPCAループヘリックスを持ち、それら領域を介してp62分子同士が多量体を形成する[13]。クライオ電子顕微鏡解析から、p62のPB1ドメイン、おそらく全長のp62も多量体化によりらせん状フィラメントを形成することがわかった[14]。一方、p62のC末端に存在するUBAドメインのX線構造解析からは、p62のUBAドメインは二量体を形成することがわかった[15]。p62のUBAドメイン同士が会合するとユビキチン鎖との親和性はほとんど無いようである。p62のUBAドメインに存在する407番目のセリン残基(S407)および403番目のセリン残基(S403)のリン酸化が、それぞれUBAドメインの構造変換およびユビキチン鎖との親和性を高める。プロテアソームの阻害などタンパク質恒常性の異常に応じて、ULK1がS407をリン酸化する(栄養飢餓では起きない)。このリン酸化は、UBAドメイン内の局所的なアミノ酸の配置を変え、p62のUBAドメイン間の相互作用が解除される[16]。また、細胞内ユビキチン量が増大するようなストレス下では、420番目のリシン残基がユビキチン化され、UBAドメイン間の相互作用を妨げるようである[17, 18]。その後、ULK1、CK2あるいはTBK1によりユビキチン結合に決定的であるMGF配列の直前に存在するS403がリン酸化を受ける。S403のリン酸化により負電荷が付加されるとユビキチン鎖との親和性が高まると想定される[19, 20]。p62がユビキチン(正確には8つのユビキチンが63番目のリシンで連結したK63鎖)と結合すると、らせん状p62フィラメントは断片化し、小さなフィラメントとなる[14]。この後の分子メカニズムの詳細は今後の課題であるが、断片化p62フィラメントとユビキチン化カーゴ複合体は小胞体近傍のオートファゴソーム形成部位に集積し、凝集体を形成する[21]。そこにAtgタンパク質が集積し、カーゴに沿って隔離膜形成が起こると考えられる[22]。最終的に、LC3やGABARAPは2つの疎水性ポケットを持っており、そこにp62のLIRのトリプトファンとロイシンが疎水性の相互作用を形成することで効率的にオートファジーにより分解される[6](図2)。これは、LC3はオートファゴソームの内外膜に局在する一方、p62の大部分はオートファゴソームの内膜に局在することと一致する[23]。
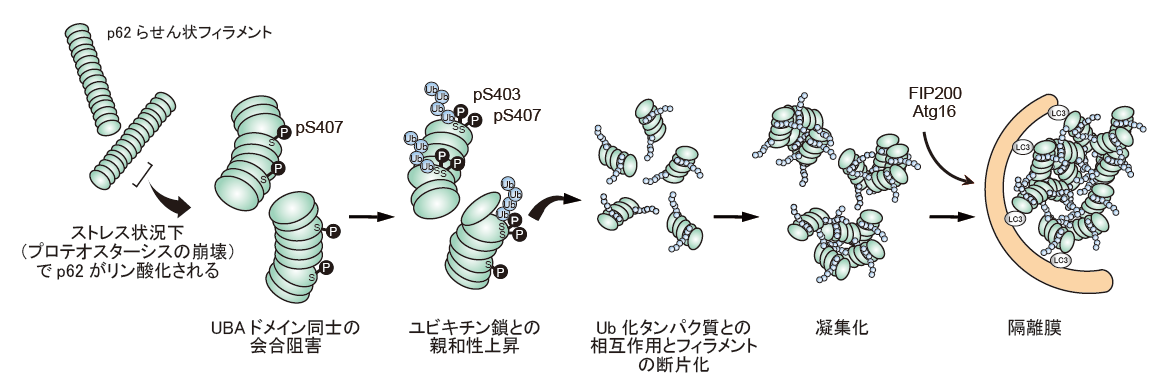
図2. p62によるユビキチン化カーゴ分解機構
プロテアソーム阻害などでプロテオスターシスが障害されるとp62のUBAドメイン内のS407がリン酸化され、UBAドメイン同士の会合が阻害される。次いで、S403がリン酸化されるとユビキチン鎖との結合親和性が上昇する。p62がユビキチン鎖と結合するとp62らせん状フィラメントは断片化し、凝集化する。その後、FIP200やAtg16などのAtgタンパク質が集積するようである。その結果として、凝集体の周辺で隔離膜が形成される。最終的に、p62とユビキチン化カーゴはp62とLC3あるいはGABARAPとの相互作用により効率的にオートファジーにより排除される。
2.p62によるNrf2活性化
Keap1-Nrf2システムにおいて、Keap1はユビキチンリガーゼ(正確にはCullin3型ユビキチンリガーゼのアダプタータンパク質)として働き、Nrf2は転写因子として生体防御酵素群の遺伝子発現を調節する[24-26]。即ち細胞が活性酸素や親電子性物質などのストレスに曝されると、Keap1がセンサーとして働き、Nrf2の分解を停止して、Nrf2が活性化する。Nrf2は核内へ移行して、抗酸化タンパク質や抗炎症性酵素の遺伝子発現を誘導し細胞を保護する。Nrf2のKeap1によるユビキチン化には蝶番と閂モデルが提唱されている[27]。Nrf2のNeh2ドメインと呼ばれる領域にあるDLGex領域とETGE配列それぞれがKeap1のβ-プロペラ構造の底面にある同じ領域に結合する[28,29]。1分子のNrf2がKeap1ホモダイマーにより認識されるのである。この2箇所の結合はNrf2のDLGex領域とETGE配列の間に存在するリシン残基のユビキチン化に不可欠である。蝶番として機能するETGE配列はKeap1と強固に結合し、閂として働くDLGex領域の結合はETGE配列に比して弱い。ストレスによりKeap1のシステイン残基が酸化修飾を受けるとDLGex領域との相互作用が解除され、Nrf2のユビキチン化が抑制されると考えられている[27](図3-A)。一方、p62はKeap1と直接に相互作用するモチーフKeap1-interactingregion(KIR)を有する[8]。KIRは、LIRの直下に存在する344から356番目のKEVDPSTGELQSL配列を指す。KIRは、STGE配列を介してNrf2が結合するKeap1のβ-プロペラ構造の底面に競合的に結合する。p62のKIRとKeap1との結合様式はNrf2のETGE配列とKeap1とのそれに酷似しているが、その親和性は低い。これは、Nrf2-ETGE配列のグルタミン酸残基に対応するアミノ酸残基がKIRの場合セリン残基(S349)になっていることに起因する。著者らは、このS349がリン酸化されることを見出した[30]。このリン酸化によりKIRのKeap1への親和性は30倍以上高まり、おそらくNrf2のDLGex領域とKeap1との結合を競合的に阻害し、Nrf2の分解を抑制、Nrf2の活性化を導くと考えられる(図3-B)。S349のリン酸化はユビキチンと結合できないF408V変異体や多量体を形成できないK7AD69A変異体ではほとんど起こらないことから、断片化p62フィラメントとユビキチン化カーゴとの複合体の形成が必要と考えられる[31]。つまり、p62がユビキチンと結合した場合にのみNrf2が活性化する。Nrf2の標的遺伝子は、抗酸化タンパク質や抗炎症性酵素のみならずAtgタンパク質やプロテアソームサブユニットの遺伝子発現をも誘導し、タンパク質恒常性維持に貢献する[32,33]。p62遺伝子もNrf2の標的遺伝子の一つであることから、ポジティブフィードバック機構が存在する[10]。また、p62の多量体形成に必須なPB1ドメイン内の7番目のリシン残基がユビキチンリガーゼTRIM21によりユビキチン化される[34]。このユビキチン化はp62のプロテアソームでの分解を促進し、Nrf2活性化を抑制する。
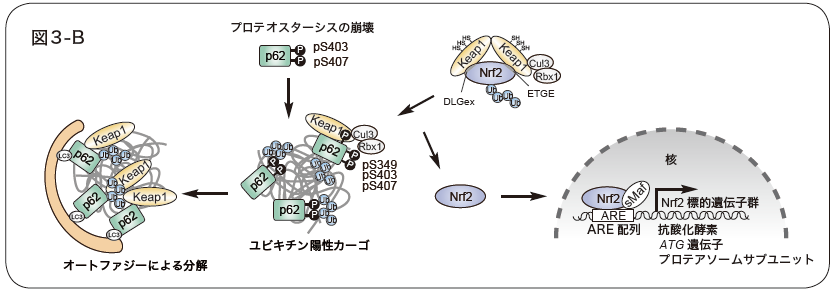
図3. Keap1-Nrf2システム
A.Nrf2は、DLGex領域、ETGE配列をKeap1ホモダイマーにより認識、ユビキチン化され、26Sプロテアソームにより分解される。親電子性物質や酸化ストレスにより、Keap1の特定のシステイン残基が酸化修飾を受けると、Nrf2との相互作用が阻害され、Nrf2は活性化する。
B.p62のUBAドメイン内の407番目と403番目のセリン残基がリン酸化(pS407、pS403)されると、ユビキチン鎖との結合親和性が高まり、p62はユビキチン化カーゴに局在化する。その後、KIRの349番目のセリン残基がリン酸化(pS349)され、p62とKeap1の親和性が高まり、Nrf2は活性化され、生体防御遺伝子群の発現が上昇する。p62は、Keap1とカーゴと共にオートファジーにより分解される。
3.p62とがん
p62はオートファジーにより選択的に分解されることから、オートファジー欠損マウス組織、特にオートファジー欠損肝臓において顕著に蓄積、凝集化する[4]。蓄積したp62は351番目のセリン残基(ヒトの349番目のセリン残基に対応)がリン酸化を受けており、Nrf2が恒常的に活性化する。このリン酸化p62を介した恒常的なNrf2の活性化が腫瘍増殖に大きな役割を果たす[30,35-37]。事実、オートファジー抑制により形成される肝腫瘍の増殖はp62ないしはNrf2を同時欠損することで大幅に抑制される[36,38]。重要なことに、ヒト肝細胞がん組織において異常蓄積が観察されている好酸性の封入体マロリー小体の主要構成成分はp62であり[39]、ヒト肝細胞がんにおいてもNrf2の活性化が予想された。実際、内在性にS349リン酸化p62を蓄積するヒト肝細胞がん株Huh-1を用いた異種移植実験から、Huh-1のp62を欠失させるとNrf2の活性化の低下とともに腫瘍の増殖が抑制されること、p62欠損Huh-1株にS349リン酸化p62模倣体を戻すとNrf2の再活性化とともに腫瘍増殖が回復することが分かった[30]。このことは、ヒト肝細胞がんでもp62を介したNrf2活性化が腫瘍増殖に寄与することを意味する。どのようにしてNrf2の活性化が肝細胞がんの増殖を助けるのであろうか?Nrf2の体細胞変異を有する(その結果として、Nrf2が恒常的に活性化している)肺腺がん細胞では、Nrf2依存的にペントースリン酸経路それに引き続くプリン核酸合成やグルタミノリシスが誘導され、それらが腫瘍増殖を支える[40]。一方、肝細胞がん細胞におけるリン酸化p62によるNrf2の恒常的活性化はグルコースからUDP-グルクロン酸の合成、そしてグルタミンからグルタチオン合成を促進させることが分かった[37]。S349リン酸化p62を持つ肝細胞がん細胞は、薬剤抱合に働くUDP-グルクロン酸およびグルタチオンの産生亢進により抗がん剤に対する耐性能を獲得し、グルタチオンの産生亢進により増殖が促進されていた(図4-A)。また、Karinらのグループは、肝細胞がん前駆細胞にp62が蓄積するとNrf2活性化だけではなく、mTORC1-c-Myc経路の活性化が起こり、これらが肝細胞がん前駆細胞から肝細胞がんへと形質転換させることを明らかにした[41](図4-B)。これらのことは、少なくとも肝細胞がんではp62は腫瘍形成にも腫瘍発達にも働くことを意味する。300例を超える日本人の肝細胞がん全ゲノムシーケンスからNrf2やKeap1の体細胞変異が特定されているが、Atg遺伝子は見つかっていない[42]。Atg遺伝子変異による肝がんの発がんは無いと言える。一方、ウイルス性肝炎や脂肪肝ではオートファジーが抑制されていることが報告されているが[43]、その結果としてp62が蓄積し、がん化あるいはがんの進展が起こっている可能性がある。
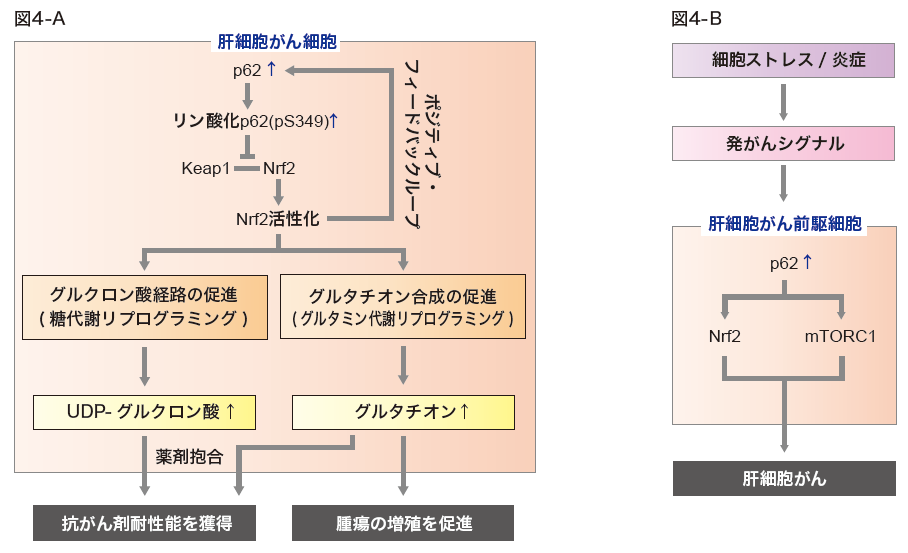
図4. 肝細胞がんにおけるp62の役割
A. 肝細胞がんにおいてp62は過剰に発現され、S349がリン酸化を受けている。その結果、恒常的にKeap1-Nrf2の相互作用が阻害され、Nrf2は継続的に活性化する。Nrf2の持続的な活性化は、グルクロン酸経路およびグルタチオン合成を促進させる。その結果、肝細胞がん細胞は増殖速度が上昇し、抗がん剤耐性能も獲得する。
B. 肝細胞がん前駆細胞においてp62が過剰に蓄積すると、Nrf2とmTORC1-c-Myc経路が活性化し、肝細胞がん前駆細胞から肝細胞がんへと形質転換させる。
おわりに
p62遺伝子変異は、骨パジェット病[44]、筋萎縮性側索硬化症や前頭側頭型認知症[45]の、p62遺伝子重複は腎明細胞がんの原因として特定されている[46]。また、昨年、幼児期に発症する遺伝性神経変性疾患においてp62遺伝子欠損が確認された[47]。これら疾患とp62のレセプターとしての、あるいはNrf2活性化タンパク質としての機能が関与するか否かを明らかにして行く必要がある。
参考文献
1. Katsuragi Y, et al.:FEBS J, 282:4672-8, 2015
2. Taniguchi K, et al.:FEBS Lett, 590:2375-97, 2016
3. Bjorkoy G, et al.:J Cell Biol, 171:603-14, 2005
4. Komatsu M, et al.:Cell, 131:1149-63, 2007
5. Pankiv S, et al.:J Biol Chem, 282:24131-45, 2007
6. Ichimura Y, et al.:J Biol Chem, 283:22847-57, 2008
7. Rogov V, et al.:Mol Cell, 53:167-78, 2014
8. Komatsu M, et al.:Nat Cell Biol, 12:213-23, 2010
9. Lau A, et al.:Mol Cell Biol, 30:3275-85, 2010
10. Jain A, et al.:J Biol Chem, 285:22576-91, 2010
11. Moscat J, et al.:Cell, 167:606-9, 2016
12. Warabi E:Seikagaku, 86:783-7, 2014
13. Lamark T, et al.:J Biol Chem, 278:34568-81, 2003
14. Ciuffa R, et al.:Cell Rep, 11:748-58, 2015
15. Isogai S, et al.:J Biol Chem, 286:31864-74, 2011
16. Lim J, et al.:PLoS Genet, 11:e1004987, 2015
17. Lee Y, et al.:Cell Rep, 19:188-202, 2017
18. Peng H, et al.:Cell Res, 27:657-74, 2017
19. Matsumoto G, et al.:Mol Cell, 44:279-89, 2011
20. Pilli M, et al.:Immunity, 37:223-34, 2012
21. Itakura E, Mizushima N:J Cell Biol, 192:17-27, 2011
22. Fujita N, et al.:J Cell Biol, 203:115-28, 2013
23. Eino A, et al.:J Cell Sci, 128:4453-61, 2015
24. Itoh K, et al.:Biochem Biophys Res Commun, 236:313-22, 1997
25. Wakabayashi N, et al.:Nat Genet, 35:238-45, 2003
26. Kobayashi A, et al.:Mol Cell Biol, 24:7130-9, 2004
27. Suzuki T, Yamamoto M:Free Radic Biol Med, 88:93-100, 2015
28. Padmanabhan B, et al.:Mol Cell, 21:689-700, 2006
29. Fukutomi T, et al.:Mol Cell Biol, 34:832-46, 2014
30. Ichimura Y, et al.:Mol Cell, 51:618-31, 2013
31. Ishimura R, et al.:FEBS Lett, 588:822-8, 2014
32. Kwak MK, et al.:Mol Cell Biol, 23:8786-94, 2003
33. Pajares M, et al.:Autophagy, 12:1902-16, 2016
34. Pan JA, et al.:Mol Cell, 61:720-33, 2016
35. Inami Y, et al.:J Cell Biol, 193:275-84, 2011
36. Takamura A, et al.:Genes Dev, 25:795-800, 2011
37. Saito T, et al.:Nat Commun, 7:12030, 2016
38. Ni HM, et al.:J Hepatol, 61:617-25, 2014
39. Zatloukal K, et al.:Am J Pathol, 160:255-63, 2002
40. Mitsuishi Y, et al.:Cancer Cell, 22:66-79, 2012
41. Umemura A, et al.:Cancer Cell, 29:935-48, 2016
42. Fujimoto A, et al.:Nat Genet, 48:500-9, 2016
43. Ueno T, Komatsu M:Nat Rev Gastroenterol Hepatol, 14:170-84, 2017
44. Singer FR:Nat Rev Endocrinol, 11:662-71, 2015
45. Rea SL, et al.:Exp Cell Res, 325:27-37, 2014
46. Li L, et al.:Cancer Cell, 24:738-50, 2013
47. Haack TB, et al.:Am J Hum Genet, 99:735-43, 2016