

国立病院機構大阪医療センター 田中 聡司 先生・
大阪大学 吉森 保 先生より 『脂肪肝病態形成における
オートファジー制御蛋白Rubiconの役割』
田中 聡司 先生
国立病院機構大阪医療センター消化器内科
【略歴】
| 平成17年3月 | 大阪大学医学部医学科 卒業 |
| 平成17年4月 | 大阪府立病院機構大阪急性期・総合医療センター 前期研修医 |
| 平成19年4月 | 大阪府立病院機構大阪急性期・総合医療センター消化器内科 後期研修医 |
| 平成22年4月 | 大阪大学医学部附属病院消化器内科 医員 |
| 平成29年3月 | 大阪大学大学院医学系研究科医学専攻 博士課程修了 |
| 平成29年4月 | 国立病院機構大阪医療センター消化器内科 医員 |

吉森 保 先生
大阪大学大学院医学系研究科
生化学・分子生物学講座遺伝学教室
脂肪肝病態形成におけるオートファジー制御蛋白Rubiconの役割
非アルコール性脂肪性肝疾患(NAFLD)は過量の栄養摂取が原因となる生活習慣病であり、近年患者数は増加傾向にある。未だ有効な薬物療法はなく、一部の症例で重症化し肝硬変、肝がんへと進展することが問題であり、病態解明が望まれる。今回我々は、過量脂質摂取に伴うRubicon発現増加がオートファジー機能を低下させ、NAFLD病態形成の一因となることを明らかにした。オートファジーが過量脂質摂取という環境因子により変容し、病態形成に寄与することが初めて示されたので概説する。
1.はじめに
非アルコール性脂肪肝は肝細胞内に脂肪滴が蓄積した状態であり、過栄養に伴う内蔵脂肪蓄積やインスリン抵抗性を背景として発症し、メタボリック・シンドロームの肝病変と考えられている。脂肪肝の原因としてはアルコール性と非アルコール性に大別され、アルコール摂取量がエタノール換算で男性は1日30 g未満(女性は1日20 g未満)で脂肪肝の場合を非アルコール性脂肪性肝疾患(nonalcoholic fatty liver disease: NAFLD)とよぶ。近年、NAFLDは最も多くみられる慢性肝疾患であり、先進国では人口の約30%が罹患し増加傾向にある。NAFLDのうち約10%は炎症所見や肝線維化を伴う非アルコール性脂肪肝炎(nonalcoholic steatohepatitis: NASH)へと進展し、肝硬変・肝がんを発症することが問題となっている[1, 2]。現在、NAFLDに有効な薬物療法はなく、病態進展予防および有効な治療法の確立には病態解明が必要である。
オートファジーは細胞内成分を分解するシステムで、オルガネラや蛋白などの細胞構成成分を対象にライソゾームへと輸送し分解へ導き、細胞内の不要物を分解することにより浄化作用を持つ。これまで、培養細胞や動物を用いた検討において、NAFLD/NASH病態形成環境下でオートファジーは抑制されているという現象が報告されてきた[3, 4]。近年、オートファジーが肝細胞内で脂質を選択的に分解し、脂肪代謝を制御しているという報告がなされlipophagyと名付けられた[3]。これはNAFLD病態形成において、過栄養に伴う肝細胞内への脂質流入により生じた脂肪滴蓄積が、リポファジーの抑制により更に増悪する可能性を示唆するものであったが、そのメカニズムは未解明であった。
今回我々は高脂肪食負荷の際に、肝細胞においてオートファジーを負に制御するRubiconの発現上昇によるオートファジー抑制が、NAFLDの病態進展に関与していることを明らかにした[5]。本稿では、この研究について概説したい。
2.培養肝細胞株を用いた脂肪酸負荷時におけるオートファジー抑制の検討
パルミチン酸を添加した培養肝細胞(HepG2)では、細胞内での脂肪滴蓄積を伴って細胞死(アポトーシス)が誘導された。また、オートファジーの評価を行うと、パルミチン酸添加によりLC3-Ⅱ発現量は増加すること、およびオートファジー阻害剤(Bafilomycin)投与時のLC3-Ⅱ発現量の変化を指標とするオートファジー・フラックスは低下することから、オートファゴソーム形成後の段階でのオートファジー抑制が示唆された。
オートファジー抑制因子を検討するために、オートファジー関連タンパク質の発現量をオートファジーの進行段階別に評価した。まずオートファジーの開始を負に制御するmTOR経路は抑制されており、またオートファゴソーム形成を制御するAtg5、Atg7およびBeclin1の発現に変化を認めなかったことから、これらのタンパク質はオートファジー抑制には寄与していなかった。次に、オートファゴソームとリソソームの融合を負に制御するRubiconに着目すると、その発現が増強していた(図1A)。オートファジー制御蛋白Rubicon(Run domain Beclin-1 interacting And cysteine-rich containing protein)は、Beclin1と結合する蛋白として発見され、オートファジーの最終段階であるオートファゴソームとライソゾームの融合を抑制し、細胞内のRubiconが増加するとオートファジー機能が低下することを我々は報告した[6]。
パルミチン酸添加によるRubicon増加の意義を検討するためにsiRNAを用いてRubiconをノックダウンしたところ、オートファジー抑制が解除されたことから、パルミチン酸投与によるRubicon増加がオートファジー抑制の原因であると考えられた(図1B)。Rubiconのノックダウンによりオートファジー抑制を解除すると、パルミチン酸投与下での細胞内脂肪滴蓄積量は減少し、アポトーシスも軽減した。また、パルミチン酸投与時のRubicon増加のメカニズムに関しては、遺伝子発現量は変化を認めず、パルスチェイス・アッセイの結果からRubicon分解が遅延することにより蛋白量が増加することが示された。よってRubiconの分解遅延・発現増強を介したオートファジー抑制が、パルミチン酸添加による脂肪滴蓄積を増加させ、アポトーシスを誘導していることが示された。
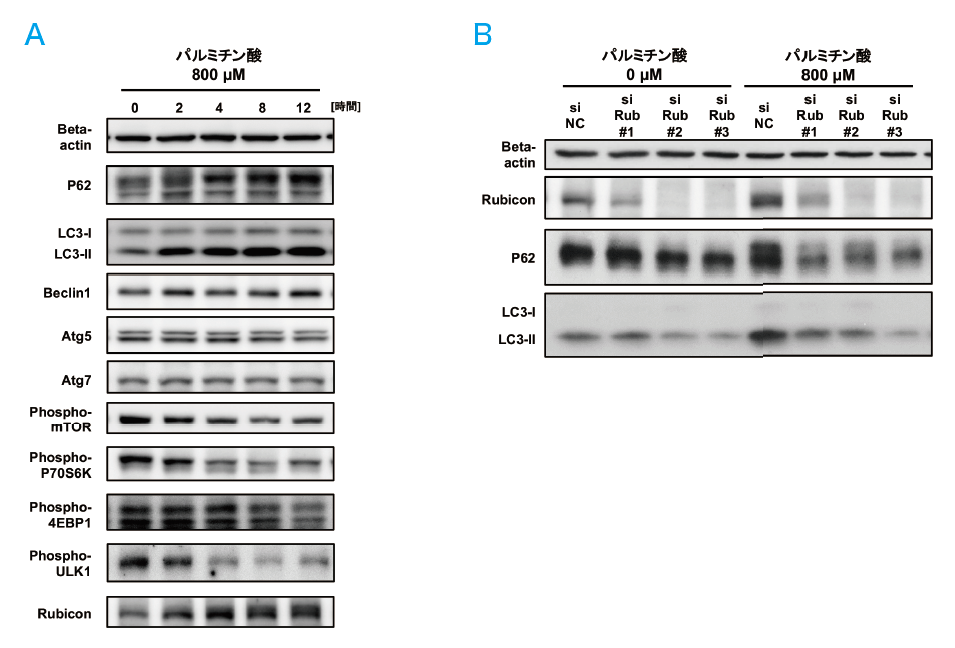
図1. 培養肝細胞株HepG2へのパルミチン酸投与によりRubicon発現増加を介したオートファジー抑制を認めた
(文献5より引用)
A.パルミチン酸投与によりRubicon発現増加を認めた。
B.Rubiconをノックダウンすると、パルミチン酸投与によるP62およびLC3-Ⅱの発現増加は抑制され、
オートファジー抑制が軽減した。
3.マウスNAFLDモデルにおけるオートファジー抑制の検討
次に、NAFLDモデルとして野生型マウスに高脂肪食を4ヶ月摂取させて検討した。このモデルでは、摂取2ヶ月後から肝臓内に脂肪滴が蓄積し、4ヶ月では脂肪肝に伴う著明な肝腫大を認め、ALTは300 IU/L程度にまで上昇する。高脂肪食摂取4ヶ月の時点での肝臓ではP62の発現増強を認め、その遺伝子発現量に変化を認めなかったことから、オートファジー抑制が示唆された。またLC3-Ⅱ発現は増強しており、電子顕微鏡で観察するとオートファゴソームが増加していたことから、オートファゴソーム形成後の段階でのオートファジー抑制が示唆された。培養細胞の実験と同様にオートファジー抑制因子を検討すると、mTOR経路は抑制されており、Atg5、Atg7、Beclin1の発現に変化を認めなかったが、Rubiconは高脂肪食投与1ヶ月後から発現増強していた。
そこで肝細胞特異的Rubiconノックアウト(KO)マウス(Alb-Cre Rubicon fl/fl)を作製した。このマウスは発生・発育に異常を認めず、肝細胞においてオートファジーが亢進している他は生理的条件下では表現型を示さなかった。野生型マウスおよびRubicon KOマウスに高脂肪食を4ヶ月摂取させ評価した。Rubicon KOマウス肝臓では野生型マウスに比してP62ならびにLC3-Ⅱ発現量は減少しており、高脂肪食摂取に伴うオートファジー抑制が改善していた(図2A)。また肝細胞内脂肪滴蓄積量および肝臓内中性脂肪含有量、肝腫大はいずれもRubicon KOマウスで軽減しており(図2B)、電子顕微鏡観察において、Rubicon KOマウスでは、脂肪滴の辺縁に膜様構造物の集積を認め、リポファジーが亢進している可能性が示唆された(図2C)。更に、ALT値ならびに肝組織TUNEL染色陽性細胞数はRubicon KOマウスで低下しており、アポトーシスが抑制されていた。よって、マウスNAFLDモデルにおいてもRubiconの発現増強を介したオートファジー抑制が、脂質過量摂取に伴う肝臓内脂肪蓄積を増悪させ、アポトーシスを誘導していることが示された。
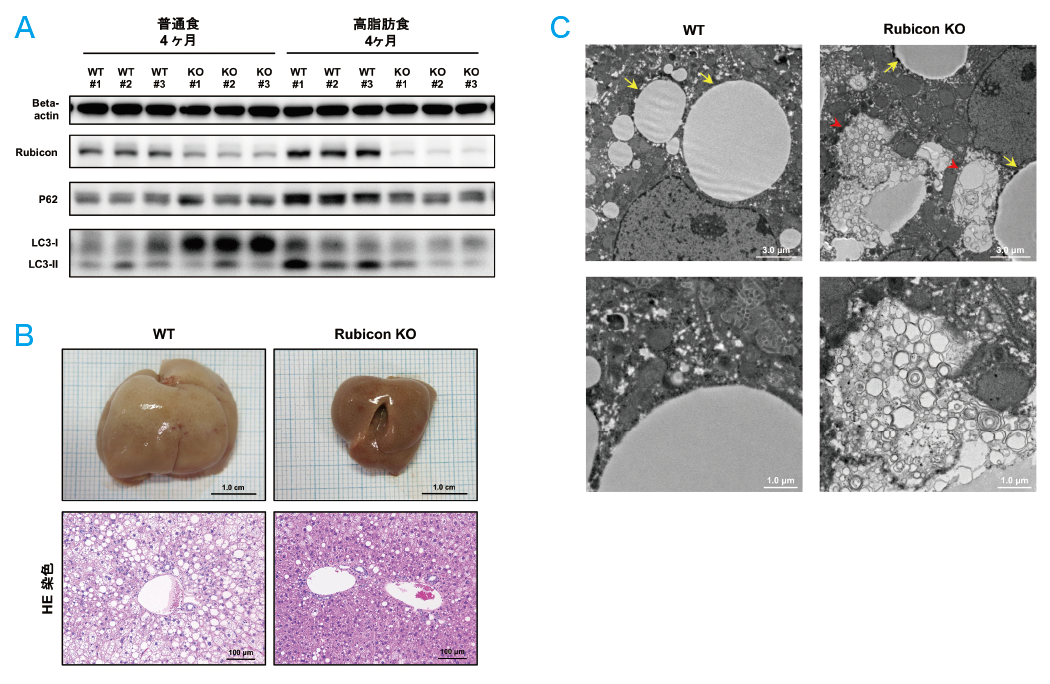
図2. 肝細胞特異的Rubicon KOマウスに高脂肪食を摂取させると、野生型マウスに比してオートファジー抑制は
軽減し、脂肪肝は改善した(文献5より引用)
A.高脂肪食摂取に伴うP62およびLC3-Ⅱの発現増加は、Rubicon KOマウスで軽減した。
B.高脂肪食摂取に伴う肝腫大および肝細胞内脂肪滴蓄積は、Rubicon KOマウスで軽減した。
C.高脂肪食を摂取したRubicon KOマウス肝臓を電子顕微鏡で観察すると、脂肪滴辺縁に膜様構造物の蓄積を認め、
リポファジー亢進が示唆された。(矢印は脂肪滴、矢頭は膜様構造物を指す)
4.ヒト切除肝におけるRubicon発現の検討
ヒトNAFLD患者においてもRubiconの発現増加が認められるかを検討するために、肝切除手術検体を用いて検討した。肝切除術を受けた患者で、ウイルス性肝炎既往がなく、アルコール消費量がエタノール換算で20 g/日を満たす者の肝組織HE染色所見で脂肪肝の有無別に分け、Rubiconの発現量をウエスタンブロットで評価した。HE染色で肝脂肪滴を認める症例では、肝脂肪滴を認めないに比し、Rubicon発現が増加していた(図3)。これより、ヒトNAFLD症例においてもRubicon発現上昇を介した病態形成の可能性が示唆された。
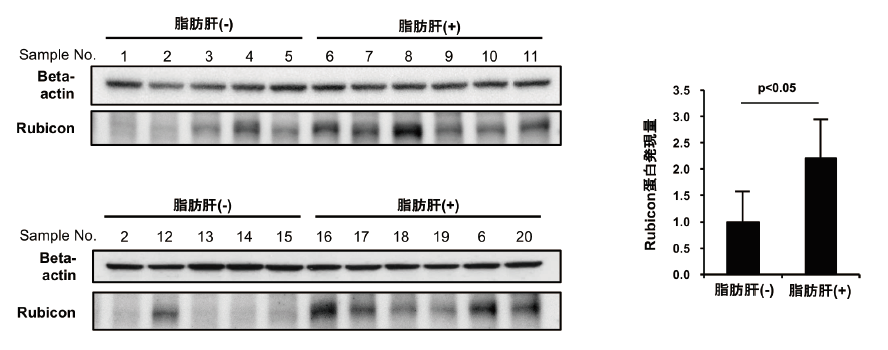
図3. ヒトNAFLD肝臓ではRubicon発現が増加していた(文献5より引用)
5.おわりに
近年オートファジーに関する研究は飛躍的に進展しており、様々な疾患の病態形成に関与していることが示されている。生活習慣病であり、近年患者数が増加している非アルコール性脂肪性肝疾患(NAFLD)の病態形成において、過量脂質摂取に伴うRubicon発現増加を介したオートファジー機能低下が一因であることが本研究により示された(図4)。オートファジーが過量脂質摂取という環境因子により変容し、病態形成に寄与することを初めて報告した。いまだ有効な薬物治療法が存在しないNAFLDに対して、Rubiconを標的としたオートファジーの亢進が、肝内脂肪蓄積を減少させ肝細胞アポトーシスを抑制する治療法の開発にもつながる可能性がある。更に肝細胞アポトーシスの抑制は、肝硬変への進展や肝がんの発症を予防することにもつながる効果が期待される。
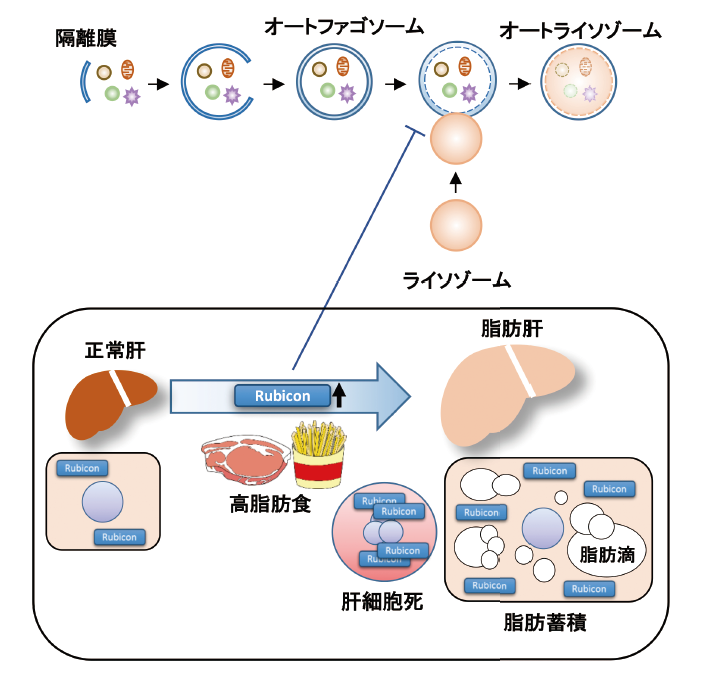
図4. 高脂肪食摂取は肝細胞内のRubiconを増加させてオートファジーを抑制し、脂肪滴分解抑制および細胞死誘導
を促進する
参考文献
- Demir M, Lang S, Steffen HM. Nonalcoholic fatty liver disease-current status and future directions.
J Dig Dis. 2015; 16(10): 541-557. - Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013; 10(11): 686-690.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009; 458(7242): 1131-1135.
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010; 11(6): 467-478.
- Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016; 64(6): 1994-2014.
- Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009; 11(4): 385-396.