

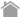
Articles from key opinion leaders
"p62/Sqstm1 : The molecule that links autophagy to the Keap1-Nrf2 system"

|
 |
|
| Dr. Masaaki Komatsu Department of Biochemistry, Niigata University Graduate School of Medical and Dental Sciences |
Dr. Yoshinobu Ichimura Department of Biochemistry, Niigata University Graduate School of Medical and Dental Sciences |
p62/Sqstm1: The molecule that links autophagy to the Keap1-Nrf2 system
Autophagy and the Keap1-Nrf2 system are cell defense mechanisms induced in response to environmental stress. Cells are protected by autophagy via the degradation of cytotoxic cellular components in the lysosome, and by the Keap1-Nrf2 system via the induction of expression of antioxidant proteins. Although the two pathways are induced by common stress conditions, their interaction is not known. Recently, p62/Sqstm1, an Nrf2 target gene product that is selectively degraded by autophagy, was identified as a key molecule that links the two pathways.
Introduction
p62/Sqstm1 (hereinafter referred to as p62) is a multifunctional, stress-inducible protein containing a Phox and Bem1p (PB1) domain, zinc finger, two nuclear localization signals, a TRAF6-binding domain, nuclear export signal, an LC3-interacting region (LIR), a Keap1-interacting region (KIR), and a ubiquitin-associated domain (UBA) [1, 2] (Fig. 1). p62 is conserved in metazoan organisms and has been studied as a signaling adapter protein for atypical PKC, ERK1, NF-κB, caspase-8, and mTORC1 since its discovery. Since 2005, p62 has become widely recognized after Johansen and colleagues proposed its function as a receptor that brings ubiquitinated protein aggregates to autophagosomes [3]. We reported in 2007 that p62 directly interacted with the autophagosomal protein LC3, and that the accumulation of p62 was marked and that p62 significantly accumulated and formed aggregates with ubiquitinated proteins in tissues of autophagy-deficient mice [4]. Furthermore, the biochemical analysis performed by Johansen and colleagues in 2007 and the structural analysis performed by the authors in 2008 demonstrated that p62 interacted with LC3 via the DDDWTHL sequence (LIR) [5, 6]. The biochemical and structural studies of LIR in p62 laid the groundwork for numerous autophagy receptors and LC3/Atg8-binding proteins now known to contain an LIR. In the following years, several studies demonstrated that p62 functioned as a receptor that recognized depolarized mitochondria and intracellular bacteria via ubiquitin chains and transported these cargos to autophagosomes [7]. In 2010, independent studies by our group and Zhang’s group revealed a mechanism wherein p62 directly interacted with Keap1, which is an adaptor protein for a Cullin-3-type ubiquitin ligase, leading to the activation of Nrf2, the master transcription factor for oxidative stress response [8, 9]. Furthermore, Johansen and colleagues showed that p62 was an Nrf2 target gene, indicating a positive feedback mechanism for the p62-Keap1-Nrf2 pathway [10]. Thus, p62 has been on the center stage of research on autophagy and oxidative stress response since 2005. This article will review the current knowledge regarding the mechanism of Nrf2 activation in association with the function of p62 as an autophagy receptor. Readers are encouraged to refer to other reviews regarding the function of p62 as a signaling hub [11, 12].
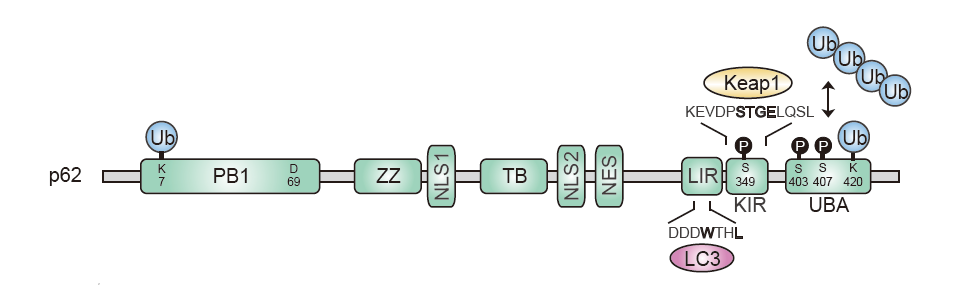
Figure 1. Domain structure of human p62
p62 oligomerizes via the lysine residue at position 7 in the Phox and Bem1p (PB1) domain and the aspartic acid residue at position 69, and forms flexible spiral filaments. Ubiquitination of the lysine residue at position 7 by the ubiquitin ligase TRIM21 inhibits oligomerization. The tryptophan residue at position 340 in the LC3-interacting region (LIR) and the leucine residue at position 343 form hydrophobic interactions with the two hydrophobic pockets in LC3 or GABARAP. The phosphorylation of the serine residue at positions 407 in the Ubiquitin-associated (UBA) domain at the C-terminus induces a conformational change in the UBA domain, and the phosphorylation of the serine residue at position 403 increases the affinity for ubiquitin chains. The ubiquitination of the lysine residue at position 420 inhibits dimerization. The Keap1-interacting region (KIR) binds to Keap1. The STGE sequence from positions 349 to 352 mediates binding to the bottom surface of the β-propeller structure of Keap1. The phosphorylation of the serine residue at position 349 increases the affinity of KIR for Keap1 over 30-fold.
ZZ, zinc finger; TB, TRAF6-binding domain; NLS, nuclear localization signal; NES, nuclear export signal.
1. p62 as a receptor for selective autophagy
The PB1 domain at the N-terminus of p62 contains a cluster of basic amino acids and the OPCA loop/helix that contains a cluster of acidic amino acids. These regions mediate homooligomerization of the p62 molecule [13]. Based on cryo-electron microscopy analysis, the PB1 domain of p62, and presumably the full-length p62, oligomerize to form spiral filaments [14]. In addition, X-ray structural analyses showed that the UBA domain at the C-terminus of p62 forms a dimer [15]. When the UBA domains of p62 interact with each other, its affinity for ubiquitin chains is lost. Phosphorylation of the serine residue at position 407 (S407) in the UBA domain of human p62 induces a conformational change in the domain, and phosphorylation of the serine residue at position 403 (S403) increases the affinity for ubiquitin chains. S407 is phosphorylated by ULK1 in response to the disruption of proteostasis by proteasome inhibition (not by nutrition starvation). This phosphorylation induces localized conformational changes of amino acids within the UBA domain, which releases the interaction between the UBA domains of p62 [16]. When intracellular ubiquitin increases in response to certain stress conditions, the lysine residue at position 420 is ubiquitinated, which blocks the interaction between the UBA domains [17, 18]. Subsequently, S403 is phosphorylated by ULK1, CK2, or TBK1. S403 is located in front of the MGF sequence, which is critical for ubiquitin binding. The phosphorylation of S403 increases the negative charge, which presumably enhances the affinity for ubiquitin [19, 20]. When p62 binds to ubiquitin (more precisely, the K63 ubiquitin chain, in which 8 ubiquitin molecules are linked through the lysine at position 63), the spiral p62 filaments are fragmented and become smaller filaments [14]. Although details of the molecular mechanism of subsequent processes remain to be determined, the complex of fragmented p62 filaments and the ubiquitinated cargo accumulates at the site of autophagosome formation near the endoplasmic reticulum to form aggregates [21]. Atg proteins are recruited to this site, and an isolation membrane surrounding the cargo is formed [22]. In the final stage, the tryptophan and leucine residues in the LIR of p62 form hydrophobic interactions with the two hydrophobic pockets of LC3 or GABARAP, leading to the efficient autophagic degradation of p62 [6] (Fig. 2). This is consistent with the observation that LC3 is localized to the inner and outer membranes of autophagosomes, while most of p62 is localized to the inner membrane of autophagosomes [23].
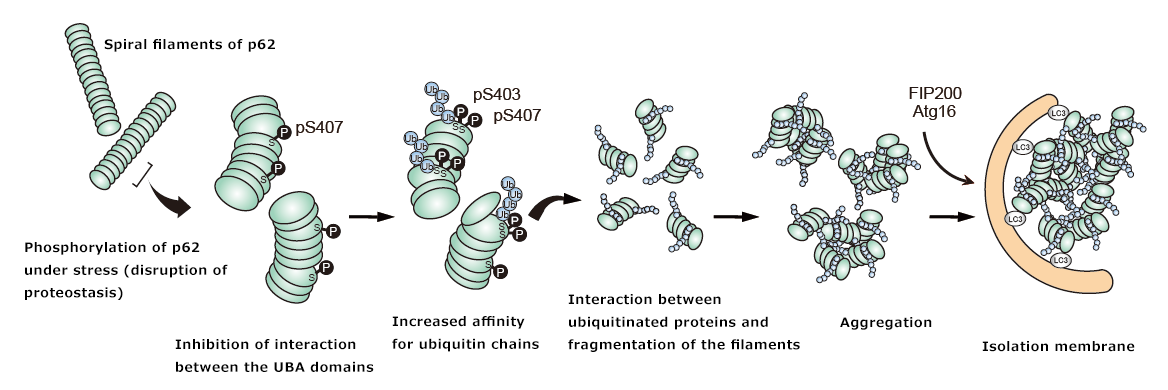
Figure 2. Mechanism of p62-mediated degradation of ubiquitinated cargo
When proteostasis is disrupted by the inhibition of the proteasome, S407 in the UBA domain of human p62 is phosphorylated, which inhibits the interaction between the UBA domains. The subsequent phosphorylation of S403 increases the affinity for ubiquitin chains. Upon binding to ubiquitin chains, the spiral filaments of p62 become fragmented and aggregated. Subsequently, Atg proteins, such as FIP200 and Atg16, are thought to be recruited, leading to the formation of an isolation membrane around the aggregates. Eventually, p62 and the ubiquitinated cargo are efficiently degraded by autophagy via the interaction between p62 and LC3 or GABARAP.
2. Nrf2 activation by p62
In the Keap1-Nrf2 system, Keap1 functions as an ubiquitin ligase (more precisely, as an adaptor protein for a Cullin-3-type ubiquitin ligase). Nrf2 is a transcription factor that regulates the expression of genes for cellular defense enzymes [24-26]. When the cell is exposed to stress conditions, such as reactive oxygen species and electrophilic compounds, Keap1 acts as a sensor and inhibits the degradation of Nrf2, resulting in the activation of Nrf2. Nrf2 translocates to the nucleus and induces the expression of genes for antioxidant proteins and anti-inflammatory enzymes to protect the cell. The “hinge and latch” model has been proposed for the ubiquitination of Nrf2 by Keap1 [27]. The DLGex region in the Neh2 domain of Nrf2 and the ETGE sequence of Nrf2 each bind to the same region of Keap1 located at the bottom surface of the β-propeller structure [28, 29]. Thus, one molecule of Nrf2 is recognized by a Keap1 homodimer. This two-site binding step is essential for the ubiquitination of the lysine residue located between the DLGex region and the ETGE sequence of Nrf2. The ETGE sequence, acting as a hinge, strongly binds to Keap1, while the binding of the DLGex region, acting as a latch, is weaker than that of the ETGE sequence. Stress-induced oxidative modification of cysteine residues of Keap1 releases its interaction with the DLGex region, and the ubiquitination of Nrf2 is prevented [27] (Fig. 3A). p62 also has a Keap1-interacting region (KIR) that directly interacts with Keap1 [8]. The KIR has the sequence KEVDPSTGELQSL, from position 344 to 356 immediately downstream of LIR. KIR competitively binds to the bottom surface of the β-propeller structure of Keap1 where Nrf2 binds via the STGE sequence. The mechanism of interaction between the KIR of p62 and Keap1 is nearly the same as that between the ETGE sequence of Nrf2 and Keap1; however, the binding affinity is low. This is because the amino acid corresponding to the glutamine in the Nrf2-ETGE sequence is serine (S349) in KIR. We found that S349 can be phosphorylated [30]. The phosphorylation of S349 increases the affinity of KIR for Keap1 more than 30-fold, which presumably competes with the DLGex region of Nrf2 for Keap1 binding and inhibits the degradation of Nrf2, leading to the activation of Nrf2 (Fig. 3B). The phosphorylation of S349 is largely blocked in F408V mutants, which cannot bind ubiquitin, and in K7A D69A mutants, which cannot form oligomers. These observations indicate that the complex formation between fragmented p62 filaments and the ubiquitinated cargo is required for S349 phosphorylation [31]. In other words, Nrf2 activation occurs only when p62 binds to ubiquitin. Nrf2 target genes are not limited to antioxidant proteins and anti-inflammatory enzymes. Nrf2 also induces the expression of genes for Atg proteins and proteasome subunits, thereby contributing to proteostasis [32, 33]. Because p62 gene is one of the Nrf2 target genes, a positive feedback mechanism exists [10]. Additionally, the lysine residue at position 7 within the PB1 domain, which is essential for the oligomerization of p62, is ubiquitinated by the ubiquitin ligase TRIM21 [34]. This ubiquitination facilitates the proteasomal degradation of p62 and inhibits Nrf2 activation.
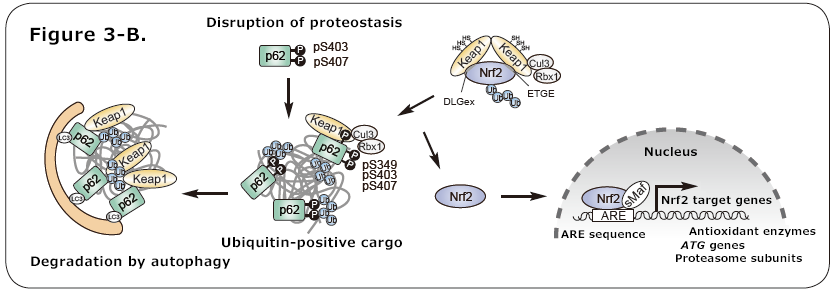
Figure 3. The Keap1-Nrf2 system
A. The DLGex region and ETGE sequence of Nrf2 are recognized by Keap1 homodimers and are ubiquitinated, leading to degradation by the 26S proteasome. When specific cysteine residues of Keap1 undergo oxidative modification due to exposure to electrophilic compounds or oxidative stress, the interaction with Nrf2 is inhibited, leading to the activation of Nrf2.
B. When the serine residues at position 407 and 403 in the UBA domain of p62 are phosphorylated (pS407, pS403), the affinity for ubiquitin chains increases and p62 becomes localized to the ubiquitinated cargo. Subsequently, the serine residue at position 349 in KIR is phosphorylated (pS349), which increases the affinity of p62 for Keap1, leading to the activation of Nrf2 and the induction of expression of cellular defense genes. p62 is degraded by autophagy along with Keap1 and the cargo.
3. p62 and cancer
p62 is selectively degraded by autophagy, and thus marked accumulation and the formation of aggregates in tissues of autophagy-deficient mice are observed, especially in autophagy-deficient livers [4]. Accumulated p62 is phosphorylated at the serine residue at position 351 (corresponding to the serine residue at position 349 in humans), and Nrf2 is constitutively activated. The constitutive activation of Nrf2 via phospho-p62 plays a significant role in tumor growth [30, 35-37]. In fact, the growth of liver tumors caused by the inhibition of autophagy is greatly diminished by concomitant deletion of p62 or Nrf2 [36, 38]. More importantly, p62 is a major constituent of Mallory bodies, which are an eosinophilic inclusion known to abnormally accumulate in human hepatocellular cancer tissues [39], suggesting the activation of Nrf2 in human hepatocellular cancers. A human hepatocellular carcinoma cell line, Huh-1, intrinsically accumulates phospho-S349 p62. Xenograft experiments with Huh-1 cells demonstrate that the deletion of p62 in Huh-1 cells reduced Nrf2 activation and tumor growth. The forced expression of a phosphorylation-mimic allele of p62 (S349E) in p62-deficient cells restored Nrf2 activation and tumor growth [30]. These findings indicate that Nrf2 activation via p62 contributes to tumor growth in human hepatocellular cancer. How does Nrf2 activation facilitate the growth of hepatocellular cancer? Lung adenocarcinoma cells with somatic mutations in Nrf2 (and resulting constitutively active Nrf2) exhibit Nrf2-dependent activation of the pentose phosphate pathway and its downstream purine nucleotide synthesis and glutaminolysis, which all support tumor growth [40]. In hepatocellular cancer cells, the constitutive activation of Nrf2 by phospho-p62 facilitates UDP-glucuronic acid synthesis from glucose and glutathione synthesis from glutamine [37]. Hepatocellular carcinoma cells with phosphorylated p62 (pS349) acquired resistance against anticancer drugs through the increased production of glutathione and UDP-glucuronic acid, which are involved in drug conjugation. The increased production of glutathione also promoted tumor growth (Fig. 4A). A study by Karin’s group demonstrates that accumulation of p62 in hepatocellular carcinoma progenitor cells activates not only Nrf2, but also the mTORC1-c-Myc pathway, which induces the transformation of progenitor cells into hepatocarcinoma [41] (Fig. 4B). These findings indicate that p62 is involved in tumorigenesis, as well as tumor development, at least in hepatocellular carcinoma. The whole genome sequencing of over 300 hepatocellular carcinoma tissues from Japanese patients revealed somatic mutations in Nrf2 and Keap1; however, Atg mutations were not found [42]. Thus, mutations in Atg genes are unlikely to cause liver cancer. On the other hand, autophagy is inhibited by viral hepatitis and hepatic steatosis [43], which could lead to p62 accumulation and consequent tumorigenesis and tumor progression.
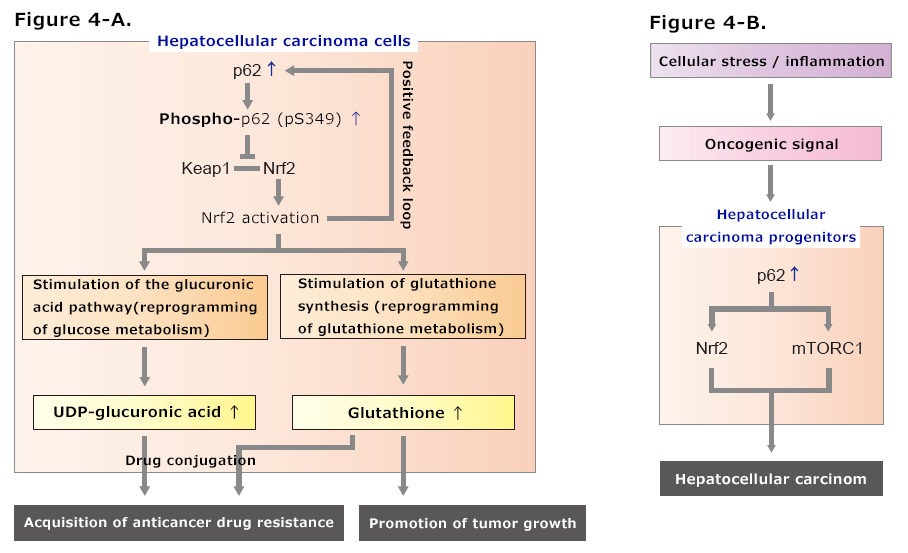
Figure 4. Role of p62 in hepatocellular carcinoma
A. p62 is overexpressed, and S349 is phosphorylated in hepatocellular carcinoma. As a result, Keap1-Nrf2 interactions remain inhibited and Nrf2 is constitutively activated. The constitutive activation of Nrf2 stimulates the glucuronic acid pathway and glutathione synthesis, resulting in the promotion of growth of hepatocellular carcinoma cells and the acquisition of anticancer drug resistance.
B. In hepatocellular carcinoma progenitors, the excessive accumulation of p62 leads to the activation of Nrf2 and the mTORC1-c-Myc pathway, resulting in transformation of the progenitors into hepatocellular carcinoma.
Conclusions
Mutations in the p62 gene have been identified as being one of the causes of Paget's disease of bone [44] and amyotrophic lateral sclerosis and frontotemporal lobar degeneration [45], and the amplification of the p62 gene is one of the causes of clear cell renal cell carcinoma [46]. More recently, p62 gene deficiencies have been identified in childhood-onset hereditary neurodegeneration [47]. Further studies are warranted to clarify whether p62 is involved in these diseases as a receptor or as an Nrf2-activating protein.
References
1. Katsuragi Y, et al.:FEBS J, 282:4672-8, 2015
2. Taniguchi K, et al.:FEBS Lett, 590:2375-97, 2016
3. Bjorkoy G, et al.:J Cell Biol, 171:603-14, 2005
4. Komatsu M, et al.:Cell, 131:1149-63, 2007
5. Pankiv S, et al.:J Biol Chem, 282:24131-45, 2007
6. Ichimura Y, et al.:J Biol Chem, 283:22847-57, 2008
7. Rogov V, et al.:Mol Cell, 53:167-78, 2014
8. Komatsu M, et al.:Nat Cell Biol, 12:213-23, 2010
9. Lau A, et al.:Mol Cell Biol, 30:3275-85, 2010
10. Jain A, et al.:J Biol Chem, 285:22576-91, 2010
11. Moscat J, et al.:Cell, 167:606-9, 2016
12. Warabi E:Seikagaku, 86:783-7, 2014
13. Lamark T, et al.:J Biol Chem, 278:34568-81, 2003
14. Ciuffa R, et al.:Cell Rep, 11:748-58, 2015
15. Isogai S, et al.:J Biol Chem, 286:31864-74, 2011
16. Lim J, et al.:PLoS Genet, 11:e1004987, 2015
17. Lee Y, et al.:Cell Rep, 19:188-202, 2017
18. Peng H, et al.:Cell Res, 27:657-74, 2017
19. Matsumoto G, et al.:Mol Cell, 44:279-89, 2011
20. Pilli M, et al.:Immunity, 37:223-34, 2012
21. Itakura E, Mizushima N:J Cell Biol, 192:17-27, 2011
22. Fujita N, et al.:J Cell Biol, 203:115-28, 2013
23. Eino A, et al.:J Cell Sci, 128:4453-61, 2015
24. Itoh K, et al.:Biochem Biophys Res Commun, 236:313-22, 1997
25. Wakabayashi N, et al.:Nat Genet, 35:238-45, 2003
26. Kobayashi A, et al.:Mol Cell Biol, 24:7130-9, 2004
27. Suzuki T, Yamamoto M:Free Radic Biol Med, 88:93-100, 2015
28. Padmanabhan B, et al.:Mol Cell, 21:689-700, 2006
29. Fukutomi T, et al.:Mol Cell Biol, 34:832-46, 2014
30. Ichimura Y, et al.:Mol Cell, 51:618-31, 2013
31. Ishimura R, et al.:FEBS Lett, 588:822-8, 2014
32. Kwak MK, et al.:Mol Cell Biol, 23:8786-94, 2003
33. Pajares M, et al.:Autophagy, 12:1902-16, 2016
34. Pan JA, et al.:Mol Cell, 61:720-33, 2016
35. Inami Y, et al.:J Cell Biol, 193:275-84, 2011
36. Takamura A, et al.:Genes Dev, 25:795-800, 2011
37. Saito T, et al.:Nat Commun, 7:12030, 2016
38. Ni HM, et al.:J Hepatol, 61:617-25, 2014
39. Zatloukal K, et al.:Am J Pathol, 160:255-63, 2002
40. Mitsuishi Y, et al.:Cancer Cell, 22:66-79, 2012
41. Umemura A, et al.:Cancer Cell, 29:935-48, 2016
42. Fujimoto A, et al.:Nat Genet, 48:500-9, 2016
43. Ueno T, Komatsu M:Nat Rev Gastroenterol Hepatol, 14:170-84, 2017
44. Singer FR:Nat Rev Endocrinol, 11:662-71, 2015
45. Rea SL, et al.:Exp Cell Res, 325:27-37, 2014
46. Li L, et al.:Cancer Cell, 24:738-50, 2013
47. Haack TB, et al.:Am J Hum Genet, 99:735-43, 2016