
- Japan(Japanese / English)
- Global
- MBL TOP
- MBL site search

Articles from key opinion leaders
Role of the autophagy regulator Rubicon in the pathogenesis of fatty liver disease
|
 |
|
| Dr. Satoshi Tanaka Department of Gastroenterology and Hepatology, National Hospital Organization Osaka National Hospital |
Dr. Tamotsu Yoshimori Department of Genetics, Osaka University Graduate School of Medicine |

Nonalcoholic fatty liver disease (NAFLD) is a lifestyle-related disease caused by excessive nutrient intake, and the number of NAFLD patients has been increasing in recent years. Effective treatment has yet to be developed, and in some cases, the disease progresses to serious conditions such as cirrhosis and liver cancer. Therefore, there is a need to elucidate the pathogenesis of NAFLD. Recently, we demonstrated that upregulation of Rubicon (Run domain Beclin-1-interacting and cysteine-rich domain-containing protein), in association with excessive fat intake, impaired autophagy and played a pathogenic role in NAFLD. Below is an overview of the first report showing that autophagy is altered by excessive fat intake, which is an environmental factor, and thereby contributes to NAFLD pathogenesis.
1. Introduction
NAFLD is a condition in which lipid droplets accumulate in hepatocytes. It is considered a hepatic manifestation of metabolic syndrome that accompanies overnutrition-induced visceral fat accumulation and insulin resistance. Fatty liver disease is classified as alcoholic or nonalcoholic, based on the cause. The disease is called NAFLD if patients consume less than 30 g of alcohol per day for men, or less than 20 g of alcohol per day for women. In recent years, NAFLD has become the most prevalent chronic liver disease, affecting about 30 percent of the population of developed countries, and the number of patients is increasing. Approximately 10 percent of NAFLD cases progress to nonalcoholic steatohepatitis (NASH), involving inflammation and hepatic fibrosis, and could lead to cirrhosis and liver cancer [1, 2]. Currently, there are no effective therapeutic drugs for NAFLD. To prevent progression of the disease and establish effective treatment options, the pathogenesis of NAFLD needs to be elucidated.
Autophagy is a process that degrades intracellular components. Cellular constituents, such as organelles and proteins, are transported
to the lysosome for degradation, thereby ridding the cell of unnecessary materials. Previous studies in cultured cells and animals have reported that autophagy is impaired under conditions that induce NAFLD/NASH [3, 4]. More recently, autophagy was found to selectively degrade lipids
within hepatocytes to regulate fat metabolism in a process that was named “lipophagy” [3]. This finding suggested the possibility that during NAFLD pathogenesis, suppression of lipophagy might exacerbate the accumulation of lipid droplets formed upon lipid influx into hepatocytes
due to overnutrition. The mechanism, however, remained unknown.
In our study, we have demonstrated that the negative regulator of autophagy, Rubicon, is upregulated in hepatocytes under high-fat diet, which impairs autophagy and contributes to the pathogenesis of NAFLD [5]. This article provides an overview of the study.
2. Suppression of autophagy by fatty acid supplementation in a hepatocyte cell line
Palmitate supplementation in cultured hepatocytes (HepG2) induced accumulation of intracellular lipid droplets and apoptosis. Assessment of autophagy revealed that expression of LC3-II was increased with palmitate supplementation. Autophagy flux was reduced when measured by changes in LC3-II expression level in the presence
of the autophagy inhibitor Bafilomycin. These results suggest that autophagy was suppressed after the formation of autophagosomes.
To investigate autophagy suppressors, expression levels of autophagy-related proteins in each phase of the autophagy pathway were measured. First, the mTOR pathway, which is a negative regulator of the initiation of autophagy, was inhibited by palmitate supplementation. Expression levels of the regulators of autophagosomal formation, Atg5, Atg7, and Beclin1, were unaltered. Thus, these proteins were not involved in the palmitate-induced suppression of autophagy. Next, we examined Rubicon (Run domain Beclin-1-interacting and cysteine-rich domain-containing protein), which is a negative regulator of autophagosome-lysosome fusion. The expression of Rubicon was elevated by palmitate supplementation (Figure 1A). The autophagy regulator Rubicon was identified as a Beclin1-binding protein. We reported that it suppressed autophagosome-lysosome fusion, which is the final stage of autophagy, and that autophagy was impaired when cellular Rubicon was increased [6].
The physiological significance of palmitate-induced increase in Rubicon was investigated using siRNA. Autophagy impairment was attenuated by Rubicon knockdown, indicating that palmitate-induced increase in Rubicon was the cause of the impaired autophagy (Figure 1B). When impaired autophagy was attenuated by Rubicon knockdown, palmitate-induced accumulation of lipid droplets and apoptosis were reduced. The mechanism of palmitate-induced upregulation of Rubicon was also determined. While Rubicon gene expression was unaltered, the protein level was elevated due to delayed degradation, as demonstrated by pulse-chase experiments. Together, these observations indicated that suppression of autophagy, through
upregulation of Rubicon due to delayed degradation, enhanced palmitate-induced accumulation of lipid droplets and induced apoptosis.
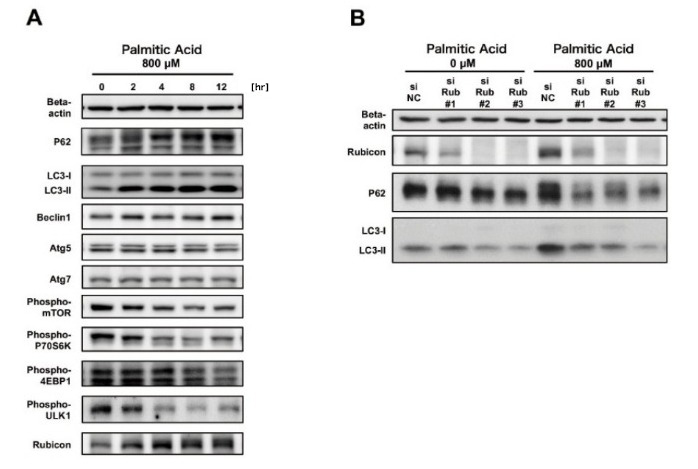
Figure 1. Autophagy was impaired through Rubicon upregulation by palmitate-supplementation in hepatocyte cell line HepG2.
A. Rubicon was upregulated by palmitate supplementation.
B. Rubicon knockdown reduced palmitate-induced upregulation of P62 and LC3-II and improved impaired autophagy.
3. Suppression of autophagy in mouse models of NAFLD
We next examined a mouse model of NAFLD in which wild-type mice were fed high-fat diet for 4 months. In this model, lipid droplets accumulated in the liver after 2 months on a high-fat diet, and marked hepatic hypertrophy with steatosis occurs after 4 months, with elevated ALT of about 300 IU/L. At 4 months on the high-fat diet, P62 was upregulated in the liver with no change in gene expression, suggesting impaired autophagy. LC3-II expression was elevated, and increased autophagosomes were observed by electron microscopy, indicating that autophagy was suppressed after autophagosome formation. To investigate negative regulators of autophagy in this mouse model, expression levels of autophagy-related proteins were examined. Western blotting analysis showed that the mTOR pathway was inhibited, and expressions of Atg5, Atg7, and Beclin1 were unaltered, while Rubicon was upregulated after 1 month on the high-fat diet. These findings raised the possibility that upregulation of Rubicon impaired autophagic flux in livers of mice on high-fat diet.
We then generated the liver-specific Rubicon knockout (KO) mice (Alb-Cre Rubicon fl/fl). These mice had no abnormalities during embryogenesis and growth, and no phenotype was observed under normal conditions except elevated autophagy in hepatocytes. Wild-type mice and Rubicon KO mice were evaluated after 4 months on the high-fat diet. Compared with wild-type mice, expression of P62 and LC3-II was reduced in the Rubicon KO mice, and the high-fat diet-induced autophagy impairment was improved (Figure 2A). Accumulation of lipid droplets in hepatocytes, neutral fat content in the liver, and hepatic hypertrophy were reduced in the Rubicon KO mice (Figure 2B). By electron microscopy, accumulation of membranous structures was observed at the perimeter of lipid droplets in the Rubicon KO mice, suggesting enhanced lipophagy (Figure 2C). Further, ALT values and TUNEL-positive cells in liver sections were lower in the Rubicon KO mice, indicating reduced apoptosis. These results show that impaired autophagy through the upregulation of Rubicon exacerbates hepatic fat accumulation upon excessive lipid intake and induces apoptosis in mouse models of NAFLD.
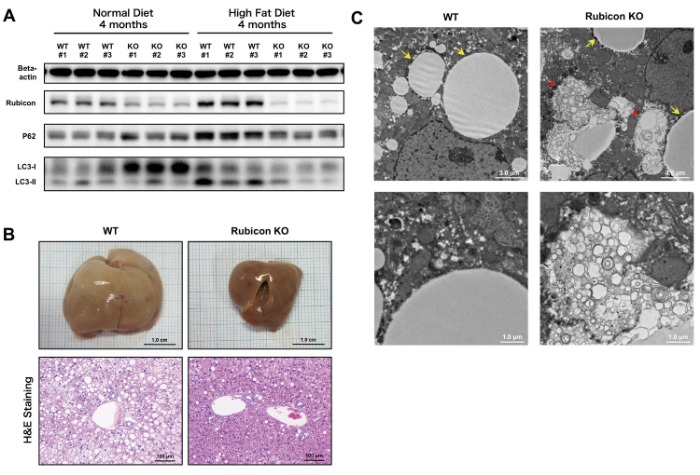
Figure 2. Impaired autophagy was attenuated, and steatosis was improved in liver-specific Rubicon KO mice on high-fat diet compared with wild-type mice on high-fat diet.
A. High-fat diet-induced upregulation of P62 and LC3-II was reduced in Rubicon KO mice.
B. High-fat diet-induced hepatic hypertrophy and accumulation of hepatic lipid droplets were reduced in Rubicon KO mice.
C. Electron microscopic observation of liver specimens of Rubicon KO mice on high-fat diet revealed membranous structures at the perimeter of
lipid droplets, suggesting enhanced lipophagy (arrows, lipid droplets; arrow head, membranous structure).Rubicon was upregulated by
palmitate supplementation.
4. Rubicon expression in resected human liver tissue
To determine whether Rubicon is upregulated in human NAFLD patients, surgical specimens of hepatectomy were examined. Tissue specimens from hepatectomy patients who had no history of viral hepatitis and consumed less than 20 g of alcohol per day were selected and grouped based on presence or absence of steatosis in H&E-stained liver tissue sections. Rubicon expression was determined by western blotting. Rubicon expression was elevated in cases with hepatic lipid droplets observed in H&E-stained sections, compared with cases without hepatic lipid droplets (Figure 3). This observation suggests the involvement of Rubicon upregulation in the pathogenesis of NAFLD in humans.
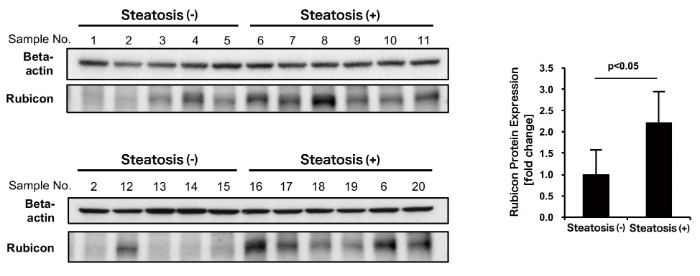
Figure 3. Rubicon was upregulated in human NAFLD liver.
5. Conclusions
Autophagy research has dramatically progressed in recent years, with recent developments showing involvement of autophagy in the pathogenesis of various diseases. NAFLD is a lifestyle-related disease with patient numbers increasing in recent years. Our study demonstrated that impaired autophagy through Rubicon upregulation, caused by excessive fat intake, played a pathogenic role in NAFLD (Figure 4). This study was the first to show that autophagy was altered by excessive fat intake, which is an environmental factor, thereby contributing to the pathogenesis of NAFLD. There is no effective treatment for NAFLD at this time, however, enhancing autophagy by targeting Rubicon could lead to the development of novel therapeutics that reduce hepatic fat accumulation and inhibit hepatocyte apoptosis. Inhibition of hepatocyte apoptosis could also prevent progression to cirrhosis and development of liver cancer.
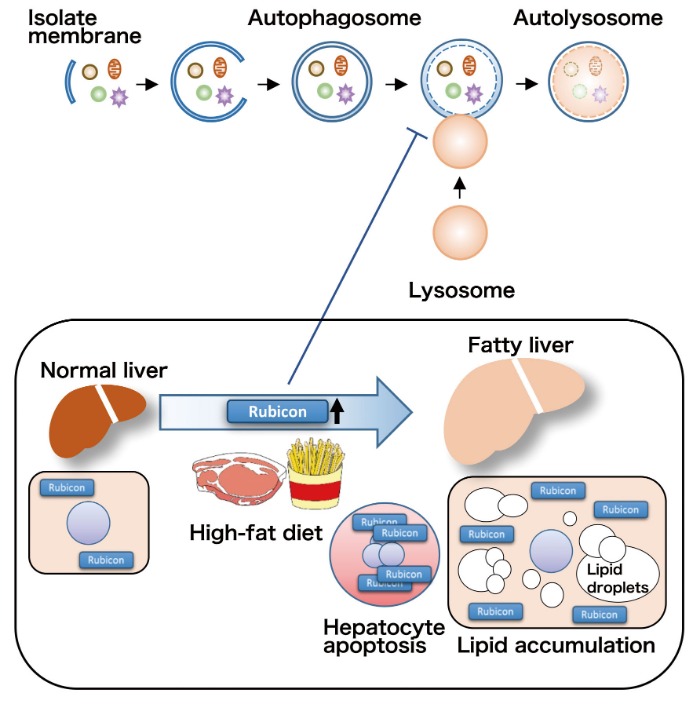
Figure 4. High-fat diet induces upregulation of Rubicon in hepatocytes, which impairs autophagy, reduces degradation of lipid droplets, and enhances induction of apoptosis.
References
1. Demir M, Lang S, Steffen HM. Nonalcoholic fatty liver disease-currentstatus and future directions.
2. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat RevGastroenterol Hepatol. 2013; 10(11): 686-690.
3. Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009; 458(7242): 1131-1135.
4. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepaticautophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010; 11(6): 467-478.
5. Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy andaccelerates hepatocyte apoptosis and lipid accumulation in nonalcoholicfatty liver disease in mice. Hepatology. 2016; 64(6): 1994-2014.
6. Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009; 11(4): 385-396.